

Abstract
Background: This comparative 52 week clinical study (NCT02213263) evaluated the efficacy, safety, immunogenicity, pharmacokinetics (PK) and pharmacodynamics (PD) of PF-05280586 (a potential rituximab biosimilar) vs. rituximab reference product sourced from the European Union (MabThera®; rituximab-EU).
Methods: Subjects with previously untreated CD20-positive, low tumor burden follicular lymphoma (LTB-FL) were randomized (1:1) to PF-05280586 or rituximab-EU (375 mg/m2 intravenously [once weekly for 4 weeks at days 1, 8, 15 and 22]). Subjects were stratified at randomization using the Follicular Lymphoma International Prognostic Index 2 (FLIPI2) classification and had an Eastern Cooperative Oncology Group performance status 0-1.
The primary endpoint was overall response rate (ORR) at Week 26, defined as the percentage of subjects achieving complete response (CR) or partial response (PR), based on central review. Therapeutic equivalence could be concluded if the 2-sided 95% confidence interval (CI) for the difference in ORR between treatment groups was within the pre-specified margin of ±16% (per US and EMA regulatory agreement). Key secondary endpoints included progression-free survival (PFS), CR rate at Week 26, time to treatment failure, duration of response, overall survival, safety, immunogenicity, PK and PD. Once all subjects completed Week 26 assessments (or early study withdrawal), the primary endpoint was evaluated.
Results: 394 subjects were randomized to PF-05280586 (n=196) or rituximab-EU (n=198); 54.8% were female, median age was 60.0 years, and 28.4% had low, 66.0% medium, and 5.6% had high risk, according to FLIPI2. 26.9% of subjects had Ann Arbor Stage II, 44.2%, Stage III, and 28.9%, Stage IV.
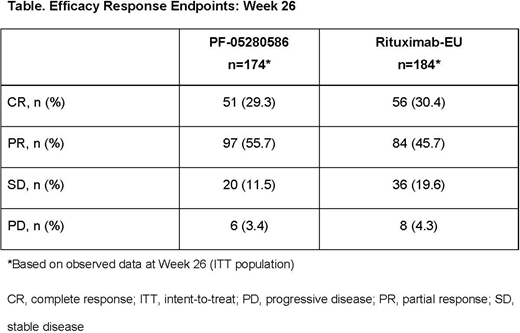
ORR at Week 26 was 75.5% (PF-05280586) vs 70.7% (rituximab-EU) for a difference of 4.66%, and the corresponding 95% CI (-4.16-13.47) was entirely contained within the pre-specified equivalence margin of ±16%. Rates of CR were 29.3% vs 30.4% in the PF-05280586 and rituximab-EU groups, respectively. At Week 26, 11.5% vs 19.6% had stable disease, and 3.4% vs 4.3% had progressive disease (PF-05280586 vs rituximab-EU, respectively) (Table). Estimated 1-year PFS rates were 76.4% (95% CI: 67.2-83.4) and 81.2% (95% CI: 72.1-87.6) in the PF-05280586 and rituximab-EU groups, respectively.
The incidence of treatment-emergent adverse events (TEAEs) (all-causality) was similar (78.6% [PF-05280586] vs 72.1% [rituximab-EU]). The most frequently reported TEAEs were infusion-related reactions (IRRs) (25.5% vs 29.9%), pruritus (6.6% vs 11.2%) and headache (8.2% vs. 9.6%) (PF-05280586 vs. rituximab-EU, respectively). The incidence of serious AEs was similar (7.7% and 6.6% in the PF-05280586 and rituximab-EU groups, respectively).
All causality grade 3 TEAEs were reported in 13.3% (PF-05280586) vs 11.7% (rituximab-EU) subjects. The most frequently reported Grade 3 TEAEs were IRR (2.6% vs 0.5%) and hypertension (1.0% vs 2.0%) in the PF-05280586 and rituximab-EU groups, respectively. The incidence of grade 3 treatment-related TEAEs was similar (PF-05280586: 13.8% vs rituximab-EU: 12.2%).
One treatment-related TEAE (grade 4 neutropenia) was reported in 1 subject in the rituximab-EU group, but was not associated with any signs or symptoms of infection. No subjects in the PF-05280586 group reported grade 4 treatment-related TEAEs. Two deaths occurred during the study (1 in each group) due to disease progression and were deemed not to be treatment-related.
Overall, 19.5% and 18.8% of subjects in the PF-05280586 and rituximab-EU groups, respectively, tested positive for antidrug antibodies (ADA) post-dose; none were positive for neutralizing antibodies. No clinically meaningful differences were observed in immune-related AEs in subjects who were treatment-emergent ADA positive vs those who were ADA negative.
Mean serum concentrations of PF-05280586 and rituximab-EU were similar, and no notable differences were observed in mean serum concentrations between ADA positive and ADA negative subjects in either treatment group. In both groups, rapid depletion in CD19 positive B-cell counts was observed after initial dosing, with recovery by week 39 and a sustained increase until end of Week 52.
Conclusions: Efficacy, safety and immunogenicity, PK and PD of PF-05280586 and rituximab-EU were similar up to Week 26 in subjects with previously untreated CD20-positive, LTB-FL.
Sharman:Pharmacyclics, an AbbVie Company: Consultancy, Research Funding; Acerta: Consultancy, Research Funding. Aurer:Pfizer: Honoraria, Research Funding; Roche: Consultancy, Honoraria, Research Funding. Robbins:Pfizer: Employment, Equity Ownership. Rosenberg:Pfizer: Employment, Equity Ownership. Khan:Pfizer: Employment, Equity Ownership. Alcasid:Pfizer: Employment.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal